
Introduction
If you are building a serum or plasma study for persisting post-acute symptoms, the fastest way to go off-track is to chase a single elevated marker and call it a day. A more reliable path is to assemble and read a long covid cytokine signature—coordinated cytokine and chemokine patterns that move together, vary by phenotype, and evolve over time. This guide translates that idea into concrete steps: what to measure first, how to design a fit-for-purpose multiplex panel (anchored in Luminex xMAP), and how to interpret results without overclaiming.
The intended readers are translational researchers and biomarker leads who must balance limited volumes, multi-batch comparability, and compliance expectations while generating decision-grade evidence. The outcome is a practical framework for target selection, panel construction, and interpretation, so you can hypothesize around immune programs rather than isolated p-values.
Why Long COVID Should Be Read as a Signature Problem
Why single markers are rarely enough
Long COVID is biologically heterogeneous across cohorts, timepoints, and symptom clusters. Individual cytokines—IL-6, TNF-α, IFN-γ, IL-8, IL-10—appear variably elevated across studies, and the effect sizes are often modest. Depending on cohort design and timing, a "significant" single-analyte shift can vanish or invert. When signals are low-grade and context-dependent, reading one marker as the whole story increases false narratives and reduces reproducibility.
Why multiplex readouts are more informative
Cytokines and chemokines typically move as coordinated programs—interferon-associated modules (IFN-γ with CXCL9/CXCL10), myeloid-recruitment axes (CCL2 with IL-6/TNF-α), or regulatory tones (IL-10, TGF-β). Signature-level interpretation is better suited for subgrouping, trajectory analysis, and hypothesis generation than isolated deltas. Multiplex datasets enable unsupervised pattern discovery and supervised confirmation in ways single-analyte readouts cannot.
What this means for study design
Panel design should reflect inflammatory axes, not a single favorite cytokine. Interpretation should prioritize biologically coherent patterns and pre-specified analytical plans over isolated p-values. Clarify whether you are running discovery, phenotype stratification, longitudinal tracking, or focused confirmation up front; each objective implies different panel breadth, QC requirements, and downstream confirmation strategy.
What to Measure First
Start from a compact, biologically coherent set that tends to be reportable in human serum or plasma. Then extend based on phenotype and study objective.
Core cytokines and chemokines
IL-6; TNF-α; IFN-γ; CXCL10/IP-10; CCL2/MCP-1; IL-8; IL-10. These span pro-inflammatory, interferon-associated, chemotactic, and regulatory axes and are a practical first pass.
Secondary or context-dependent additions
CXCL9; CCL4; IL-2; TGF-β; and optional IFN-α or CXCL11 in interferon-heavy phenotypes. Use these when the symptom structure or biological question warrants added depth.
How to prioritize targets
Prioritize reproducibility across peer-reviewed studies, expected abundance/reportability in serum or plasma, biological coherence (axes/modules), and value for phenotype separation and longitudinal tracking. A concise way to balance these factors is to keep a stable core for comparability and add phenotype-linked markers in limited tiers. The infographic and table below offer a quick reference.
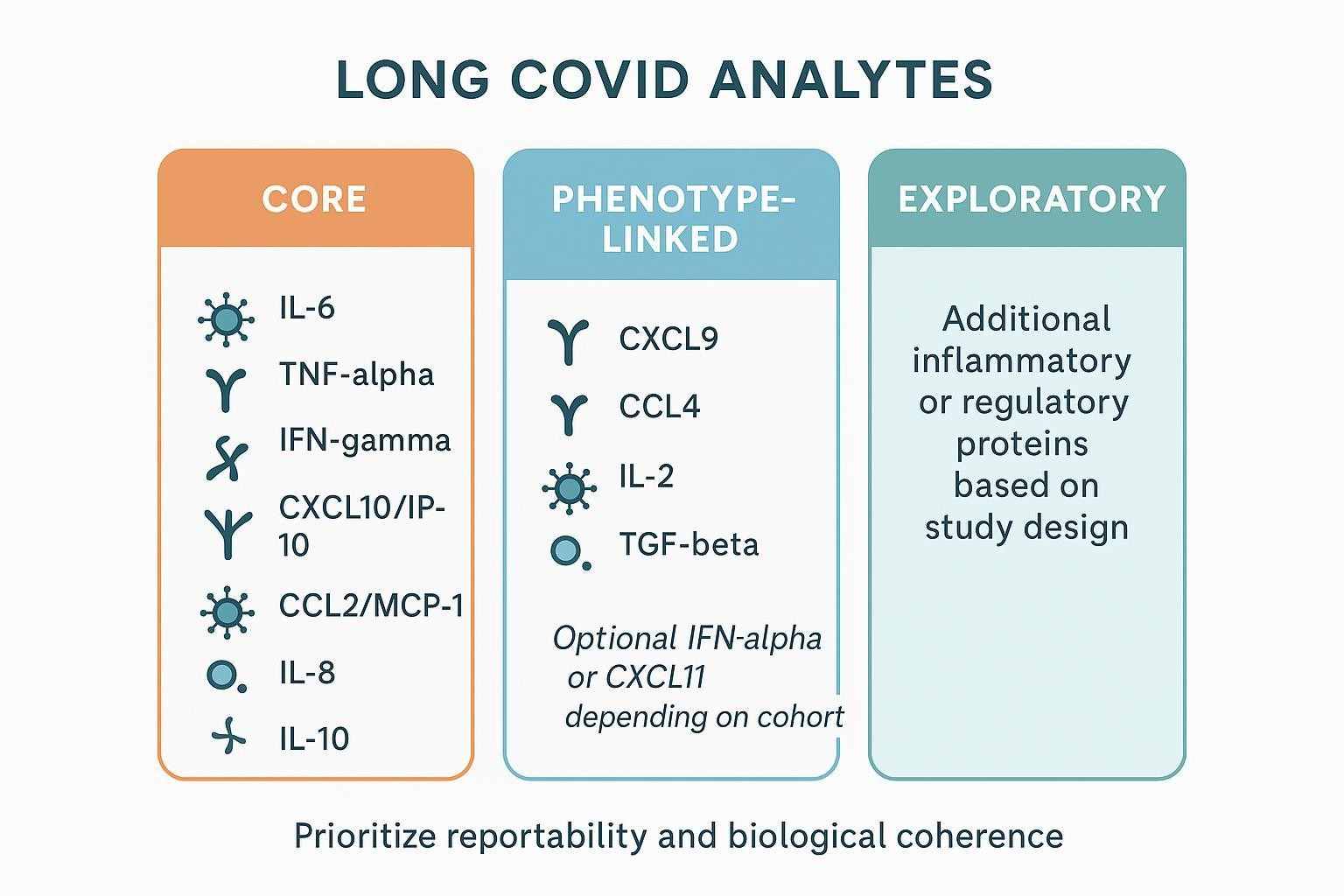
| Tier | Cytokines and chemokines | Rationale in Long COVID serum/plasma |
|---|---|---|
| Core | IL-6, TNF-α, IFN-γ, CXCL10/IP-10, CCL2/MCP-1, IL-8, IL-10 | Recurrent in literature; span pro-inflammatory, interferon-associated, chemotactic, and regulatory axes; good starting reportability |
| Phenotype-linked | CXCL9, CCL4, IL-2, TGF-β; consider IFN-α or CXCL11 | Adds depth for interferon modules, T-cell activity, regulatory tone; select based on symptom cluster hypotheses |
| Exploratory | Additional inflammatory/regulatory proteins as justified | Discovery breadth for unsupervised clustering; requires stricter downstream filtering and orthogonal confirmation |
How to Build a Fit-for-Purpose Multiplex Panel
Start from the study objective
For phenotype stratification, keep a stable set of recurrent analytes that separate clusters while limiting missingness. For longitudinal tracking, prioritize markers with reliable week-to-month dynamics and plan bridge controls. For discovery, broaden coverage but earmark a path to narrow confirmation.
Build around a stable core
Keep a core of 7–10 analytes (e.g., IL-6, TNF-α, IFN-γ, CXCL10, CXCL9, CCL2, IL-8, IL-10, plus 1–2 phenotype-specific adds) across cohorts and batches to preserve comparability. Add exploratory markers only when they serve a defined question, and predefine rules to retire low-reportability targets.
Match the panel to the sample type
Decide early between serum and plasma; maintain consistency across the cohort. Document tube type, processing time, storage, and freeze–thaw limits. When volumes are tight, plan 25–50 µL per well and avoid over-plexing if it will inflate <LLOQ frequencies.
If your workflow relies on Luminex xMAP, ensure kits and instrument settings support your concentration ranges and dynamic windows, and lock an analysis plan (5PL curves, QC acceptance limits) before first patient samples.
To see how multiplex execution and reportability work in practice, you can review the Luminex xMAP cytokine detection service and panel configuration options such as the Cytokine Panel Service . For data-processing specifics around curve fitting and QC, the Luminex Data Generation and Analysis overview may help plan acceptance criteria.
When a Broader Signature Approach Adds Value
Exploratory studies
Broader panels can reveal unexpected immune programs, particularly interferon-inducible chemokines or regulatory signals that were not in the initial hypothesis. Use unsupervised clustering to find candidate subgroups, but enforce multiple-testing control and resist overinterpreting small, noisy differences.
Stratification-focused studies
Coordinated cytokine–chemokine patterns often distinguish symptom phenotypes better than single markers. Capturing both interferon-associated (IFN-γ, CXCL9, CXCL10) and myeloid-recruitment/IL-6–TNF axes (CCL2, IL-6, TNF-α) can improve separability. Pair biology-rich metadata (symptom domains, acute severity, vaccination) with the multiplex readout to avoid confounding.
Longitudinal studies
Repeated sampling can show evolving immune trajectories as symptoms change. Stable core markers matter more than maximal plex size, and bridge controls become essential as sampling spans months. Where a signal appears to normalize over time (e.g., interferon-associated activity), ensure batch effects are not masquerading as biology.
How to Interpret Multiplex Data Without Overclaiming a Long COVID Cytokine Signature
Read patterns, not isolated p-values
Ask whether interferon-inducible chemokines move with IFN-γ, whether monocyte-recruitment signals align with IL-6/TNF-α, and whether regulatory markers shift in tandem. Treat weak, isolated signals with caution, especially near LLOQ.
Link signatures to biology carefully
Interpret coordinated changes as hypotheses about persistent inflammatory activity, myeloid recruitment, interferon-associated signaling, or regulatory/maladaptive recovery patterns. Then specify what would constitute orthogonal support (e.g., cell phenotyping, MSD confirmation of a subset, or functional assays).
Keep interpretation boundaries clear
Cytokine signatures are not standalone diagnostics. Elevation does not prove mechanism, and association with symptoms does not establish causality. Exploratory findings should be framed as hypotheses until replicated and confirmed.
What Changes Across Time, Cohorts, and Phenotypes
Time-from-infection matters
Early post-acute profiles may differ from persistent signatures months later. Some interferon-driven signals attenuate as recovery progresses, while other axes persist in subsets. Align sampling windows so you are not comparing apples to oranges across timepoints.
Phenotype structure matters
Fatigue-dominant, pulmonary, neurocognitive, or systemic presentations may not share the same immune program. Define subgrouping strategy a priori and ensure subgroup sizes support the planned analyses. Acute infection severity and treatment history can influence later signatures and should be modeled.
Cohort context matters
Control selection (pre-pandemic vs contemporary, matched comorbidities) strongly shapes interpretation. Vaccination, BMI, sex, age, and comorbidities can shift baseline levels. Cross-study comparisons require caution when cohort structures differ.
Platform and Data Strategy
Choosing the right multiplex approach
For discovery-oriented profiling and phenotype stratification, Luminex xMAP offers a balance of plex size, sample volume, and throughput appropriate for serum/plasma studies. For focused confirmation or when repeatability is paramount, MSD's electrochemiluminescence assays often provide higher sensitivity in mid-plex formats. Olink's proximity extension panels can enable broad discovery with standardized NPX units and strong cross-cohort comparability, while Simoa's digital single-molecule platform is suited to a small number of ultra–low-abundance targets when those are central to the question.
Designing around reportability
As plex size increases, the fraction of analytes below LLOQ and missing values often rises. Low-volume studies demand tighter target prioritization. It is common to perform discovery and confirmation on different platforms; plan bridging and calibration strategies if you switch.
Planning for comparability
Maintain a stable core marker set across runs or cohorts; define pre-analytical and batch rules before launch; and reserve orthogonal confirmation for the highest-value findings. For a refresher on xMAP technology and instrumentation, see What is Luminex xMAP Technology .
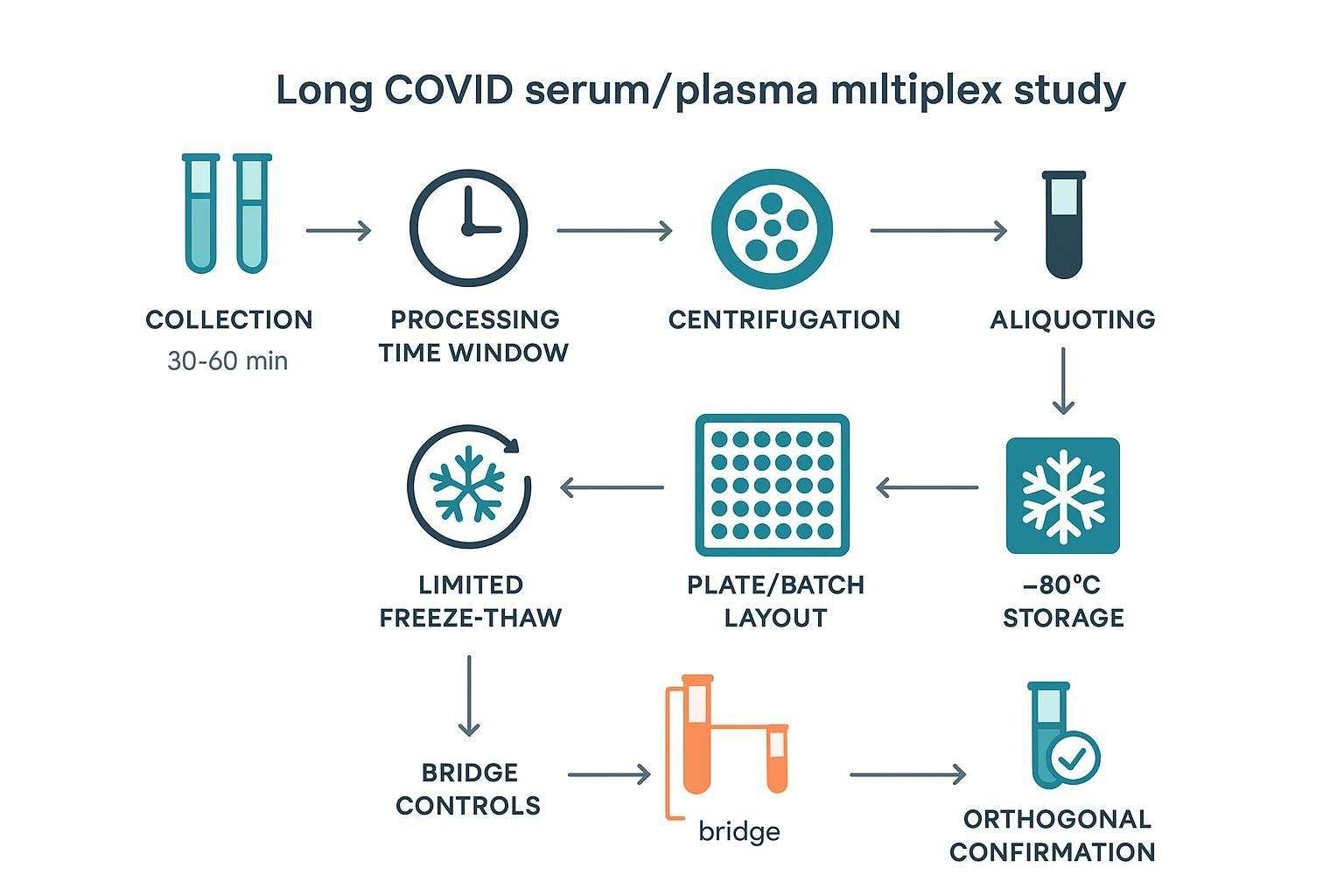
Pre-Analytical and Batch Design Considerations
Sample handling consistency
Standardize tube type (and anticoagulant if using plasma), minimize time to first spin, follow kit-specific incubation and handling guidance, aliquot promptly, and store at −80 °C with a predefined maximum number of freeze–thaw cycles. Record deviations in a sample-level log.
Batch structure
Balance symptom groups and controls across plates/runs; avoid confounding phenotype with batch; include bridge samples when longitudinal or multi-batch analyses are expected. Predefine rerun rules and %CV thresholds for acceptance.
Why this matters in Long COVID
Effect sizes are often modest. Small handling inconsistencies or batch imbalances can distort or erase low-grade inflammatory patterns. Reproducible handling is essential for interpretable signature-level data.
Common Interpretation Pitfalls and How to Correct Them
Treating one elevated cytokine as the whole story can mislead when heterogeneous biology underlies symptoms. Instead, confirm whether that analyte moves within a coherent axis (e.g., IL-6 with TNF-α and CCL2) and test the signature across phenotypes.
Building panels that are too broad for the objective increases missingness and complicates analysis. Right-size your plex for the question, pre-specify how to handle <LLOQ, and retire poorly reportable targets.
Ignoring phenotype and timepoint structure can flatten subgroup biology. Define subgroups a priori, align sampling windows, and include relevant covariates (vaccination, age, BMI) in your models.
Overinterpreting noisy low-abundance signals near the LLOQ risks false narratives. Use orthogonal confirmation (e.g., MSD for a small subset) before suggesting mechanisms.
Assuming cohorts are directly comparable overlooks differences in controls, timing, matrices, and workflows. Treat cross-study comparisons as contextual, not definitive.
FAQ
Which cytokines are most relevant in Long COVID studies?
A compact starting set includes IL-6, TNF-α, IFN-γ, CXCL10 (IP-10), CCL2 (MCP-1), IL-8, and IL-10. These span pro-inflammatory, interferon-associated, chemotactic, and regulatory axes and are commonly reportable in serum or plasma. Expand with CXCL9, CCL4, IL-2, or TGF-β when phenotype hypotheses call for added depth.
Should Long COVID studies measure chemokines as well as cytokines?
Yes. Chemokines such as CXCL9, CXCL10, and CCL2 often move with upstream cytokines and can sharpen phenotype separation. Omitting chemokines weakens signature-level interpretation and can obscure subgroup structure.
Is IL-6 enough for Long COVID biomarker profiling?
No. IL-6 can contribute to a long covid cytokine signature but is rarely sufficient by itself. Read IL-6 alongside TNF-α and chemokine modules (e.g., CXCL10, CCL2) and interpret in the context of time-from-infection and phenotype.
What is the best sample type for Long COVID cytokine studies?
Use one matrix consistently across the study. Serum is frequently chosen for multiplex cytokines due to consistency, but plasma is viable if the anticoagulant is fixed and pre-validated. The key is standardizing handling and documenting deviations.
How should multiplex data be interpreted across symptom subgroups?
Define subgroups a priori using clinical metadata, then test whether coherent axes (e.g., IFN-γ with CXCL9/CXCL10; IL-6/TNF-α with CCL2) differ between groups. Prefer multivariate approaches and adjust for multiple testing; treat subgroup signals as hypotheses until replicated.
When should a broad panel be narrowed to a focused confirmation set?
After discovery identifies a small subset that consistently separates phenotypes or tracks change over time with acceptable reportability. Move those candidates to a mid-plex confirmation platform and plan orthogonal validation before any translational claims.
References:
- Science Advances (2024): Spontaneous, persistent, T cell–dependent IFN-γ release associated with symptom persistence, with normalization paralleling recovery.
- SAGE Journals (2024): Pro-inflammatory cytokines in Long COVID, highlighting IL-6, IL-1β, and TNF-α persistence; heterogeneity across cohorts.
- Scientific Archives (2023): Elevated IL-6 in convalescents 40–93 days post-onset as a sign of ongoing activation.
- Verberk IMW et al. (2021): Pre-analytical effects on blood cytokine levels in multiplex assays; serum vs plasma considerations.
- Olink (2024): Quality and normalization concepts for multiplex immunoassays; NPX standardization overview.
- Precision for Medicine (2024): Platform overview and decision framing for biomarker programs.