
Who This Resource Is For
Common study scenarios
- Human serum or plasma cytokine studies spanning multiple plates, operators, dates, or reagent lots
- Longitudinal biomarker programs with staggered sample collection and testing timelines
- Multi-cohort or cross-project studies requiring defensible batch comparison
- Translational teams that need predefined QC and rerun rules before scaling production testing
Key decisions this guide helps you make
- What drives batch effects in multiplex cytokine panels?
- When do bridge controls become necessary—and how should they be designed?
- Which acceptance criteria should be locked before production testing?
- How should results be compared across batches or projects without overstating comparability?
- When is normalization appropriate, and when are rerun or qualification the better choice?
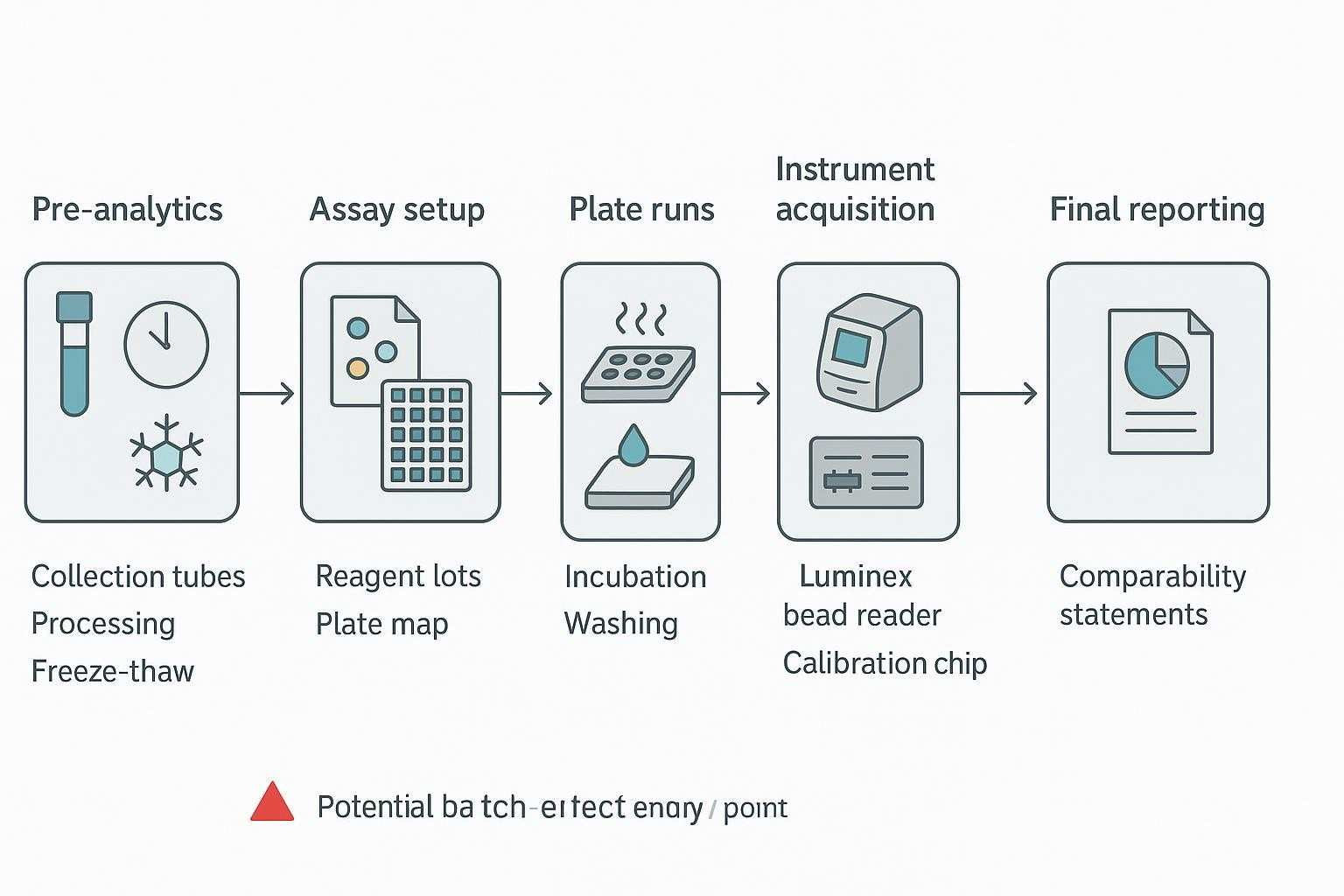
Why Batch Effects Matter in Multiplex Cytokine Studies
Batch effects are often a study-design issue, not only a data-analysis issue
Batch effects show up as technical drift—changes driven by how and when the assay was run rather than by biology. In multiplex cytokine work, that drift can hide a true treatment signal, inflate an apparent cohort difference, or create an apparent difference that weakens once plate or run structure is considered. The risk increases as studies extend across more plates, longer timelines, additional operators, or reagent lot changes, especially when the data are expected to support longitudinal interpretation, cohort comparison, or decisions that span projects.
Why this matters for decision-grade biomarker work
If a study group is confounded with a technical batch (for example, all baseline samples on one lot and all follow-up samples on another), then a statistically significant shift is not automatically a biologically meaningful one. Translational teams also need to translate QC outcomes into report language—what is reportable, what is qualified, and what cannot be pooled—so comparability depends on predefined controls and consistent review logic. When technical consistency slips, you tend to lose power, increase false discoveries, and end up with merged datasets that are difficult to defend.
Why batch discipline matters especially in Luminex-based multiplex studies
Luminex makes high-plex cytokine measurement practical at scale, which is exactly why disciplined batch planning matters. The challenge is not the platform itself, but keeping multiplex data stable, reviewable, and interpretable across time, lots, operators, and run blocks. In multi-batch Luminex studies, bridge controls, analyte-aware QC, and predefined rerun and qualification rules often determine whether observed differences can be defended as biological rather than technical.
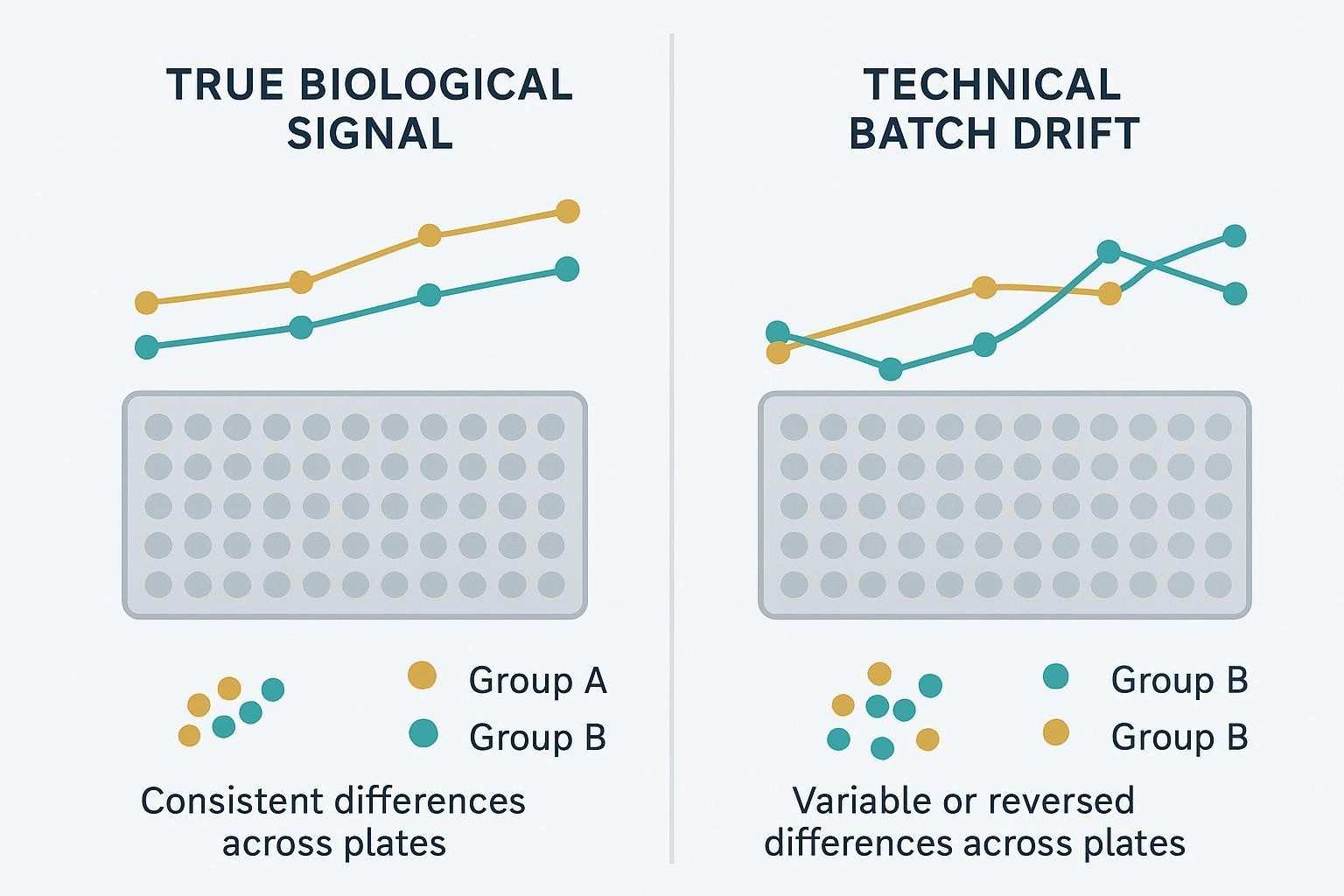
What Causes Batch Effects in Multiplex Cytokine Assays
Defining the problem
Batch effects are systematic technical differences between plates, runs, days, operators, reagent lots, instruments, or sites. The hard part is that they can look like biology—especially when the operational structure of a study overlaps with the biological structure (for example, one cohort is run early and another late, or one timepoint is tested under a different lot). The sections below group the most common sources so you can diagnose where a shift is likely coming from.
Pre-analytical sources
Many "assay problems" begin upstream. Tube type and site-specific handling can change background or recovery; processing delays and centrifugation differences can alter cell carryover and matrix interference; and uneven freeze–thaw history can selectively degrade labile cytokines. Even when handling is consistent, serum vs plasma (or different anticoagulants) can behave differently, and cross-project comparisons often inherit mismatched preprocessing decisions that were reasonable locally but incompatible globally.
Analytical sources
Within the assay itself, lot-to-lot variability is a common driver—capture/detection reagents, calibrators, and even standard-curve behavior can shift across lots. Instrument setup can drift as well (optics, detector response, calibration), and seemingly small execution differences—wash efficiency, bead recovery, incubation timing, mixing, and acquisition settings—can translate into panel-wide shifts. Operator-to-operator differences matter most when SOP details are underspecified or when teams "tune" steps over time.
Study design sources
Study design turns routine variation into high-risk bias when it is aligned with biology. If cases and controls are separated by batch, or if later timepoints are consistently processed on different days or lots, then batch identity becomes inseparable from group identity. Similar issues appear when high-priority samples are concentrated into one run block, or when exploratory and validation samples are generated under non-equivalent workflows but later compared as if they were interchangeable.
Data handling sources
Downstream rules can create or amplify batch effects even when the wet lab is stable. If plate acceptance criteria change mid-study, if rerun thresholds are applied differently across batches, or if <LLOQ, >ULOQ, and missing values are handled inconsistently, you can end up with artificial between-batch differences. Post hoc "corrections" are especially risky when they aren't tied to predefined criteria, because they can unintentionally encode the desired biological story into the data-processing step.
When Batch Effects Become a High-Risk Problem
Typical risk patterns
- Large cohorts processed over weeks or months
- Multi-center studies with uneven pre-analytics
- Longitudinal studies where later timepoints are assayed separately
- Programs evolving from exploratory profiling to decision-support use
- Projects that expect future integration with historical or follow-up datasets
Warning signs during pilot or production work
- Bridge or QC materials trend over time
- Plate-level shifts appear across many analytes at once
- Later runs show selective deterioration in certain markers
- Within-plate agreement looks acceptable, but between-plate consistency drifts
- Conclusions change depending on whether the data are grouped by biology or by batch
Questions to ask before launch
- Will all study groups be represented in every batch?
- Is there enough retained material for matrix-matched bridge controls?
- Which analytes are decision-critical and most sensitive to drift?
- Will the data be used only within one study, or also compared across studies or projects?
What Bridge Controls Are and Why They Matter
Bridge controls are shared reference materials run repeatedly across plates, batches, lot changes, or study phases to monitor technical consistency over time. Their value is not limited to showing that a single plate performed acceptably; they also provide a stable reference for judging whether data generated under changing operational conditions can still be compared in a defensible way.
| Control type | Main role | Main limitation |
|---|---|---|
| Standard curve calibrators | Define assay response and support concentration calculation | Do not reflect study-matrix behavior or endogenous sample variability |
| Kit QCs | Help judge whether a plate meets expected assay-performance criteria | May not provide a study-specific anchor for longitudinal or cross-batch comparability |
| Bridge controls | Link plates, batches, lots, or study phases through a shared reference material | Cannot fix confounded study design or make unrelated datasets automatically comparable |
Used well, bridge controls help teams:
- Detect technical drift across run date, reagent lot, operator, or acquisition conditions
- Distinguish possible technical shift from plausible biological change
- Support predefined trend review, investigation, qualification, rerun, or conditional normalization logic
- Strengthen within-study comparability and clarify the limits of cross-project comparison claims
Bridge controls are most useful when:
- Studies span multiple plates, long timelines, or lot transitions
- Longitudinal timepoints are tested in separate runs
- Cohorts or projects may later be interpreted together
- Decision-critical analytes need a stable technical anchor across production phases
Bridge controls cannot replace:
- Balanced study design across batches
- Analyte-specific review of precision, range behavior, missingness, or matrix suitability
- Predefined acceptance criteria and consistent report language
- Transparent qualification when comparability remains limited
Their value is strongest when they are tied in advance to specific decisions—such as what triggers investigation, what supports rerun, when normalization is allowed, and what level of comparison can ultimately be claimed.
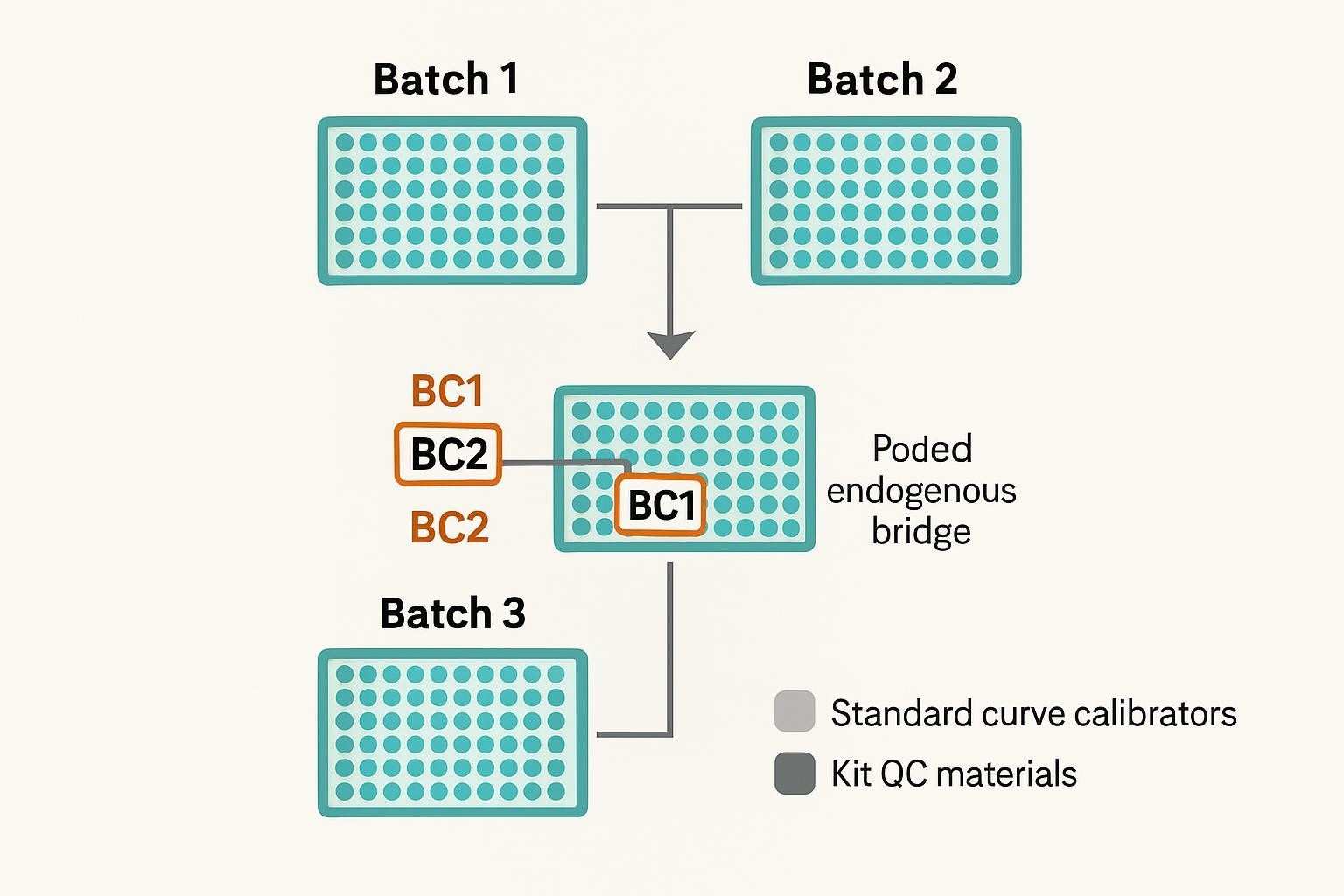
How to Design a Bridge Control Strategy
Start with the comparison goal
- Within-study batch stability
- Longitudinal comparability across timepoints
- Cross-cohort comparison
- Cross-project contextual comparison
- Formal pooled comparison with shared anchors
Choose the right bridge control material
- Match the study matrix as closely as possible
- Use material that will remain available across the full project lifecycle
- Prefer controls with usable endogenous coverage across key analytes
- Consider whether low-abundance critical targets require a spiked component
Endogenous versus spiked bridge controls
- Endogenous pooled samples are often best for reflecting matrix effects and real assay behavior
- Spiked analytes may be useful when key low-abundance cytokines are otherwise absent or below usable range
- If spiking is used, the rationale and limitations should be predefined
- Avoid turning the bridge control into an artificial material that no longer reflects study behavior
Volume planning and storage
- Estimate total bridge control volume needed for the full study
- Reserve additional contingency for repeat runs, unexpected study extension, or lot transition
- Use single-use aliquots whenever possible
- Avoid repeated freeze-thaw cycles that create artificial drift in the control itself
Decide placement and frequency
- Include bridge controls on every plate or at a predefined batch frequency
- Place them consistently enough to separate plate-position effects from batch effects
- Plan bridge placement before production begins, not after drift appears
Define what the bridge controls will govern
- Run acceptance
- Trend review
- Investigation triggers
- Rerun triggers
- Qualified normalization under predefined conditions
Acceptance Criteria: What Should Be Defined Before Study Start
A defensible cytokine program locks acceptance rules before the first production batch. The table below summarizes example categories and actions to predefine; adapt exact thresholds via pilot data and fit-for-purpose analysis.
| Level | What to check | Examples of metrics to define | Typical actions |
|---|---|---|---|
| Plate-level QC | Standard curve, QC materials, blanks, bead counts, instrument verification | Curve fit and back-calculated accuracy bands; QC recovery windows; background limits; target ≥50 beads/analyte/well; daily calibration/verification pass | Accept; investigate; rerun plate; invalidate |
| Analyte-level | Precision, range utilization, non-reportables, instability patterns | Intra-plate %CV tiers by analyte importance; %<LLOQ/%>ULOQ caps; signs of range compression; parallelism or dilution linearity where applicable | Accept; flag as descriptive; rerun subset; adjust reportability |
| Bridge control trend | Cross-plate stability, directional drift, lot/operator/date effects | Stability band vs baseline; thresholds that trigger investigation (e.g., sustained 10–20% change for decision-critical markers); analyte-aware exceptions | Accept; investigate root cause; rerun; qualify results; conditional normalization |
For Luminex-specific planning and practical execution examples, see the Luminex cytokine detection service and the Luminex multiplex assay customization tool for panel design and bridge coverage options.
Core principle
- Acceptance criteria should be locked before production testing, not written after reviewing results
- Criteria should reflect intended use, analyte importance, and tolerance for uncertainty
- Fit-for-purpose logic should guide both plate-level and analyte-level review
Plate-level quality control metrics
- Standard curve behavior
- Standard recovery consistency
- Blank or background signal review
- Bead count or acquisition sufficiency
- Overall plate validity under the selected assay workflow
Sample-level and analyte-level metrics
- Intra-assay precision where technical replicates are used
- Inter-assay behavior for bridge controls and repeated references
- Frequency of ULOQ values
- Analyte-specific missingness patterns
- Range compression or unstable performance in critical markers
Bridge-control-related acceptance logic
- What constitutes acceptable bridge control stability
- What magnitude of drift triggers review
- What conditions trigger rerun, investigation, or qualification
- When bridge-control trends are acceptable for descriptive interpretation but not for stronger comparability claims
Analyte-specific versus panel-wide rules
- Not every cytokine should necessarily be governed by the same scrutiny level
- Decision-critical analytes may require tighter review than exploratory markers
- Some rules should be assay-wide, others analyte-specific
- Uniform rules may simplify workflow but can weaken scientific defensibility
A Practical QC Framework for Multi-Batch Cytokine Panels
A workable QC framework becomes easier to run (and easier to defend) when you separate what must be true at the run level from what must be true at the analyte and study levels. The tiers below are meant to be applied in order: if Tier 1 fails, you shouldn't spend time "explaining" Tier 3 trends.
Tier 1: Run validity
Start by confirming that the run itself is technically valid: instrument status (maintenance, calibration, verification), adherence to the intended workflow and reagents, plate-level control performance, and documentation that the batch was set up according to the predefined SOP.
Tier 2: Analyte-level performance
Next, review whether each analyte behaves as expected on that plate and within that run. That includes curve behavior, precision where replicates exist, range utilization (and how often values fall outside reportable limits), plus analyte-specific missingness or instability patterns that can signal matrix effects, hook/prozone behavior, or localized execution issues.
Tier 3: Bridge control trend review
Only after the run and analytes look acceptable should you ask whether performance is stable over time. Bridge controls provide the time-linked anchor: you review cross-plate consistency, look for directional drift, and evaluate whether changes align with lot transitions, operator changes, or date-linked operational shifts.
Tier 4: Study-level comparability review
Finally, evaluate whether the dataset supports the comparison claim you want to make. That means checking whether groups were balanced across batches, whether any remaining drift overlaps with study design, whether decision-critical analytes remain interpretable across all runs, and whether the overall result should be framed as pooled, qualified analyte-level, directional-only, or descriptive-only comparison.
Best Practices for Mitigating Batch Effects Before They Happen
Strategic sample randomization
Randomization is the simplest way to prevent batch identity from becoming a proxy for biology. In practice, that means distributing cases, controls, and timepoints across plates and run dates so each batch contains a balanced mix, rather than letting operational convenience cluster one group into one run.
Reagent lot planning
If you can reserve a single lot for a large study, you remove a major source of drift. When that isn't realistic, treat lot transitions as planned events: document the change, maintain the same execution settings, and use bridge controls to quantify whether the transition produces analyte-specific shifts that need investigation or qualification.
SOP standardization
Most batch effects are "small steps repeated many times." Standardizing thawing, incubation, washing, acquisition, and review procedures reduces avoidable variation, especially when multiple operators or sites are involved. The more you need cross-batch comparability, the more those details should be explicit rather than tribal knowledge.
Predefine reporting rules early
Reporting rules are part of study design, not a cleanup step. Lock how you'll handle <LLOQ, >ULOQ, missing values, reruns, and qualification before the first production run, and make sure the QC logic can be translated into plain report language (what is comparable, what is qualified, and what should not be pooled).
How to Compare Results Across Batches Within One Study
Conditions that support comparison
- Balanced study groups across batches
- Consistent sample handling and assay workflow
- Stable bridge control behavior
- Predefined and consistently applied review rules
Situations that require caution
- Later timepoints assayed under different lots or settings
- Cases and controls separated by run schedule
- One subset of samples heavily enriched in non-reportable values
- Review criteria changed after part of the study was already completed
How to frame within-study comparability
- Comparability is earned through design, QC, and predefined decision rules
- The same panel name does not guarantee equivalent data-generation conditions
- Stronger comparison claims require stronger control of technical context
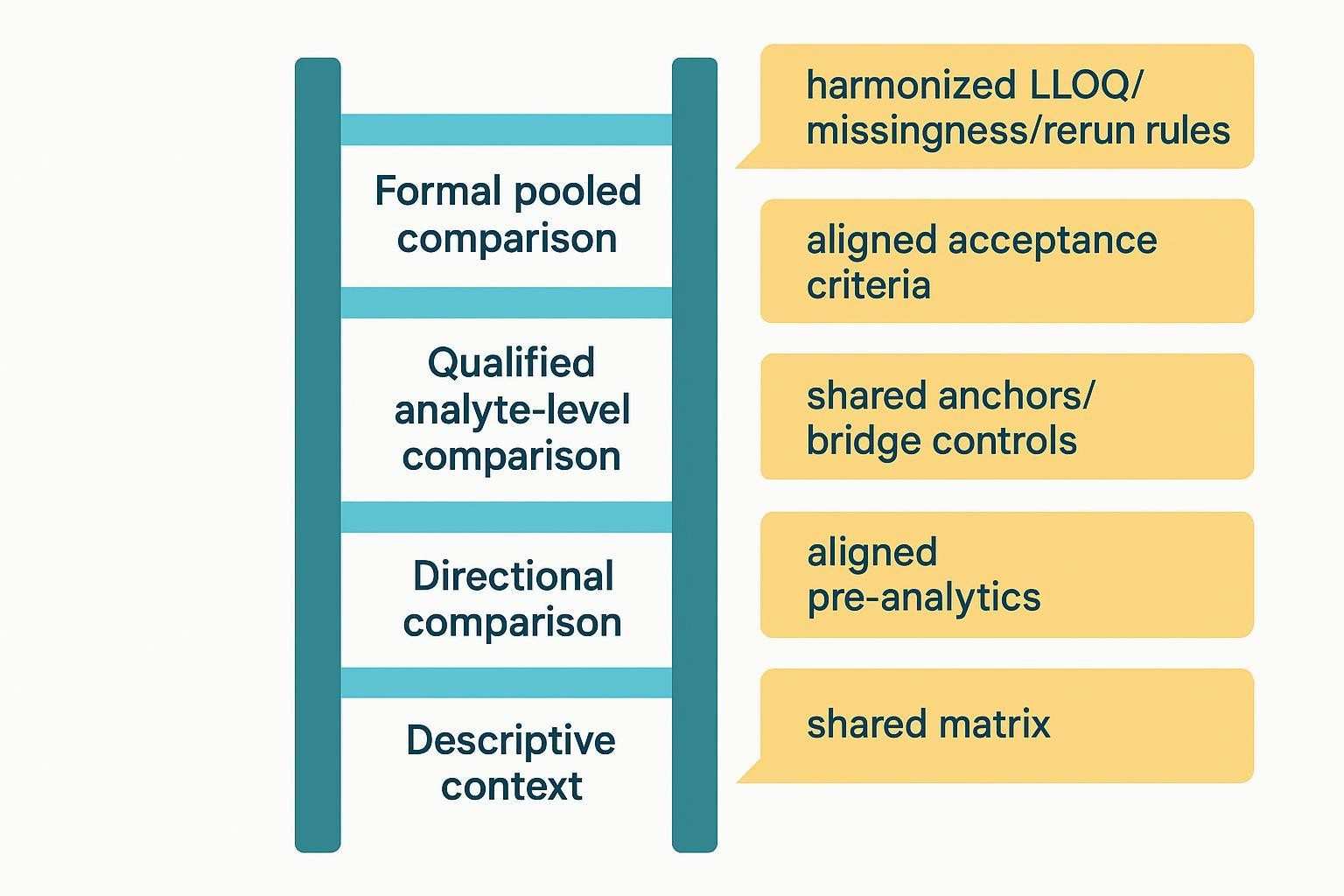
How to Compare Results Across Projects
A cross-project comparison is not binary; it's a ladder of claims earned by meeting increasing levels of preconditions. The table summarizes what each level typically allows.
| Level of comparison | Preconditions you should meet | What you can credibly claim |
|---|---|---|
| Descriptive context | Different matrices/methods; no shared anchors; exploratory objectives | Qualitative context only; note general trends without numeric linkage |
| Directional comparison | Similar matrices and workflows; stable internal QC; limited anchors | Same directionality of change across groups/cohorts; avoid precise numeric alignment |
| Qualified analyte-level comparison | Shared anchors/bridge materials; aligned LLOQ/missingness and rerun rules; cross-validated methods | Analyte-by-analyte qualified comparability with ranges/uncertainty stated |
| Formal pooled comparison | Same or cross-validated assay framework; shared bridge controls; aligned acceptance criteria and review logic | Pooled quantitative analysis with traceable, audit-ready linkage |
For human cohorts and matrix-matched panels, see the human cytokine panel service , and for inflammation or chemokine–focused marker sets vulnerable to drift, consider the human inflammation panel service and human chemokine panel service for design context.
Data Normalization: When It Helps and Where It Can Mislead
Bridge-control-based normalization
- Batch-specific correction using shared reference materials may be appropriate when predefined and well-justified
- The logic should be tied to observed bridge control behavior, not to desired biological conclusions
- Adjustment should not be used to hide unresolved design problems
Statistical normalization methods
- Mean-centering or related approaches may be considered when study design supports them
- More advanced methods can reduce some technical variance, but they rely on assumptions that may not hold in strongly confounded studies
- Statistical correction should be presented as a qualified tool, not a universal rescue strategy
MFI versus calculated concentration
- Raw signal and calculated concentration each have different strengths and limitations
- Some normalization logic may be more stable at the signal level, while final reporting often occurs at the concentration level
- The key issue is consistency, predefinition, and analyte-aware interpretation rather than one universal rule
What normalization cannot do
- It cannot fully rescue confounded study execution
- It cannot automatically make unrelated projects directly comparable
- It cannot replace transparent qualification when data remain technically limited
When to Normalize, When to Rerun, and When to Stop
Normalization is not a substitute for poor study design
- Adjustments should be predefined, limited, and justified by QC evidence
- Post hoc correction should not be used to force a preferred biological narrative
Reruns should be rule-based
- Reruns should be triggered by predefined technical criteria
- Unexpected biology alone should not justify retesting
- The rerun policy should specify which failures matter for the intended use of the data
Sometimes qualification is the more credible outcome
- Some analytes may remain descriptive only
- Some batches may support within-batch interpretation but not pooled analysis
- Transparent qualification is often more trustworthy than aggressive correction or selective rerunning
Common Mistakes to Avoid
Adding bridge controls without deciding what they govern
- Controls should be tied to acceptance, investigation, normalization, or rerun logic
Using one simplistic rule for all analytes
- Cytokines often differ too much in abundance, stability, and range behavior for one uniform rule
Treating kit QC as a full substitute for study-specific bridge controls
- Kit controls support assay validity, but may not capture matrix-matched longitudinal comparability
Waiting until drift appears to define acceptance criteria
- Retrospective rule-making weakens defensibility and increases bias
Treating cross-project comparison as automatic
- Shared biology does not eliminate technical context differences
Separating assay QC from report language
- QC decisions directly affect what can be claimed in the final report
A Practical Planning Framework Before Study Launch
Step 1: Define the comparison goal
Start by writing down the strongest claim you expect to make with the final dataset. "Within-plate consistency," "across-batch comparability," and "longitudinal comparability" often require different controls than "cross-project contextual comparison," and a formal pooled comparison is the most demanding because it requires traceable linkage across runs.
Step 2: Rank analytes by decision importance
Not every analyte should carry the same decision weight. Define which cytokines must support decisions, which are important but secondary, and which are exploratory, then align QC scrutiny and reporting language to those tiers.
Step 3: Design the bridge control plan
Specify the bridge material type (and whether it is endogenous or includes a limited spiked component), forecast the quantity you'll need with contingency, define placement and frequency, and document exactly what bridge behavior will govern during review and decision-making.
Step 4: Lock acceptance criteria
Convert the plan into rule sets that can be executed consistently: plate rules, control rules, drift rules, rerun triggers, and qualification rules. The goal is to make "what happens next" predictable when the run is good, marginal, or clearly out of bounds.
Step 5: Predefine what "comparable" means
Before any data are generated, decide how you will label outcomes. In practice, teams usually need multiple tiers—descriptive, directional, qualified analyte-level, and pooled quantitative comparison—because different analytes and batches may legitimately land in different buckets.
Pre-Study Checklist
Scientific checklist
Before you lock the assay logistics, make sure the biological comparison itself is written clearly: what are you comparing, which analytes must remain stable across batches for the interpretation to hold, and is cross-project comparison a true goal or just a possible future use case that should be treated as optional?
Analytical checklist
From an analytical perspective, finalize bridge control material and placement, define acceptance criteria, and confirm a run-review workflow that people can follow consistently. This is also the right time to align how you will treat LLOQ-related censoring, missingness, reruns, and qualification, and to decide whether any normalization options are allowed—and under what predefined conditions.
Operational checklist
Operationally, focus on removing avoidable confounding: balance sample groups across batches, plan for lot changes and instrument maintenance, reserve enough bridge material for the full study (plus contingency), standardize SOP execution across operators or sites, and document assumptions before the first production batch so you don't end up rewriting rules after seeing results.
FAQ
Do all cytokine panel studies need bridge controls?
Not always. Bridge controls are most valuable when you expect technical context to change over time—multiple plates, dates, operators, or lots—or when you want to compare across timepoints, cohorts, or projects. For a single-plate pilot used only for internal exploration, the incremental value is often low, provided plate-level QC is solid and the study won't be merged with future runs.
Can kit QC samples replace a custom bridge control?
Usually not. Kit QCs mainly tell you whether a given plate run is acceptable under the kit's QC expectations, but they may not behave like your study matrix and often aren't designed to anchor longitudinal comparability. A matrix-matched bridge control is meant to act like a repeatable "study sample" that you can trend across plates and lots.
Can bridge controls make two different projects directly comparable?
Only to a point. Bridge controls can provide a shared anchor, but strong cross-project comparability still depends on aligned matrices and pre-analytics, similar workflows, and harmonized rules for LLOQ handling, missingness, reruns, and qualification. If those conditions aren't met, the most defensible outcome may be descriptive or directional comparison rather than pooled quantitative claims.
What if no bridge control was included?
You may still be able to do limited alignment, but you should lower the strength of the claim and state the uncertainty clearly. The situation is most workable when batches were balanced by study group, internal QC was stable, and you have some overlap material (for example, repeated samples or retained aliquots) that can serve as a late-added anchor for a method-comparison style check.
Should acceptance criteria be the same for every analyte?
Usually not. Cytokines differ in abundance, stability, and susceptibility to matrix effects, so one uniform threshold can be either too strict (creating unnecessary reruns) or too loose (masking drift in decision-critical markers). A common approach is to tier analytes by decision importance and apply tighter criteria to the markers that must support conclusions.
Should normalization use MFI or calculated concentrations?
It depends on your assay framework and how you intend to report results. The key is to pick one approach in advance, justify it, and apply it consistently across batches; switching between MFI and concentrations post hoc because one "looks better" undermines interpretability and can bias downstream conclusions.
References:
- Rountree W., et al. EQAPOL multiplex program: sources of variability in bead-based cytokine assays (2012–2024). Available via PubMed Central: https://pmc.ncbi.nlm.nih.gov/articles/PMC11246216/
- Abe K., et al. Cross-platform comparison of highly sensitive immunoassays—implications for pooling and comparability (2024). PubMed Central: https://pmc.ncbi.nlm.nih.gov/articles/PMC10948291/
- Andreasson U., et al. A practical guide to immunoassay method validation (BIOMARKAPD SOPs) (2015). PubMed Central: https://pmc.ncbi.nlm.nih.gov/articles/PMC4541289/
- FDA: M10 Bioanalytical Method Validation and Study Sample Analysis (harmonized).
Looking for study-specific panel design and bridge coverage planning? The cytokine panel service provides broader context on matrices, analyte coverage, and workflow selection for longitudinal programs.